Overview
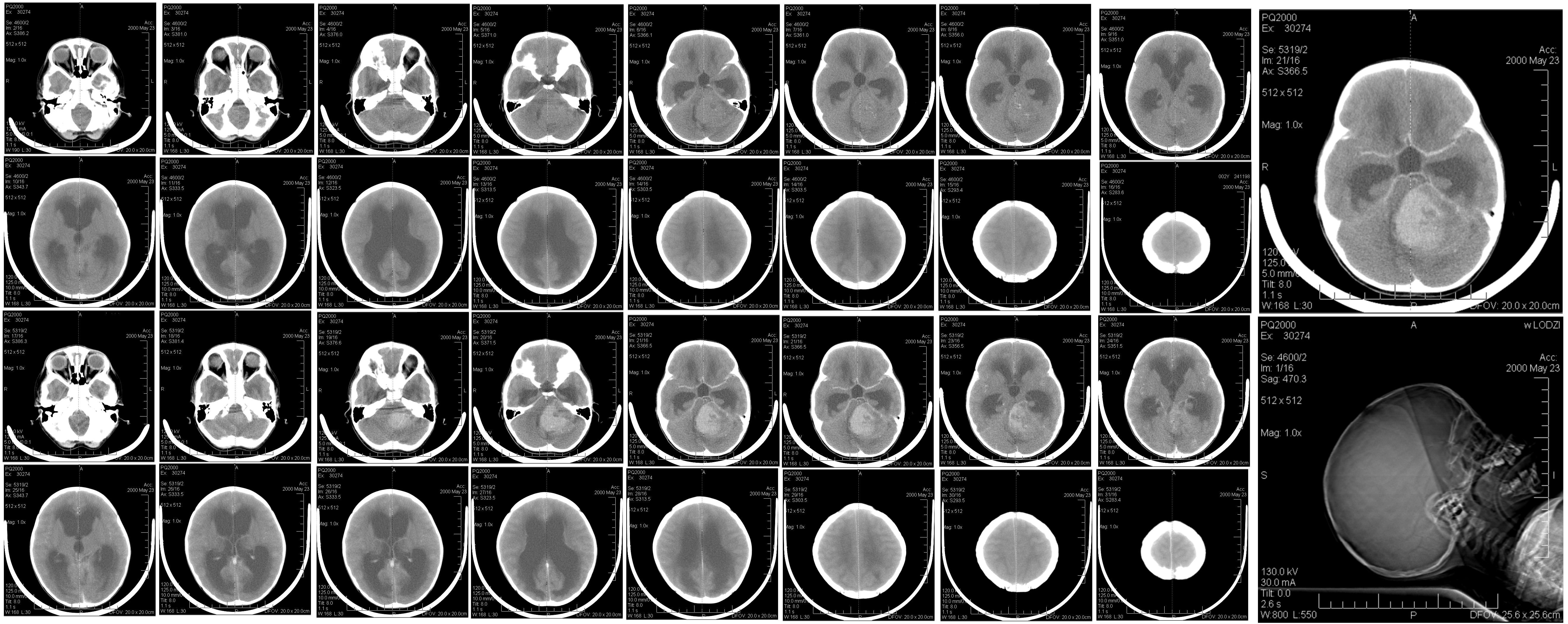
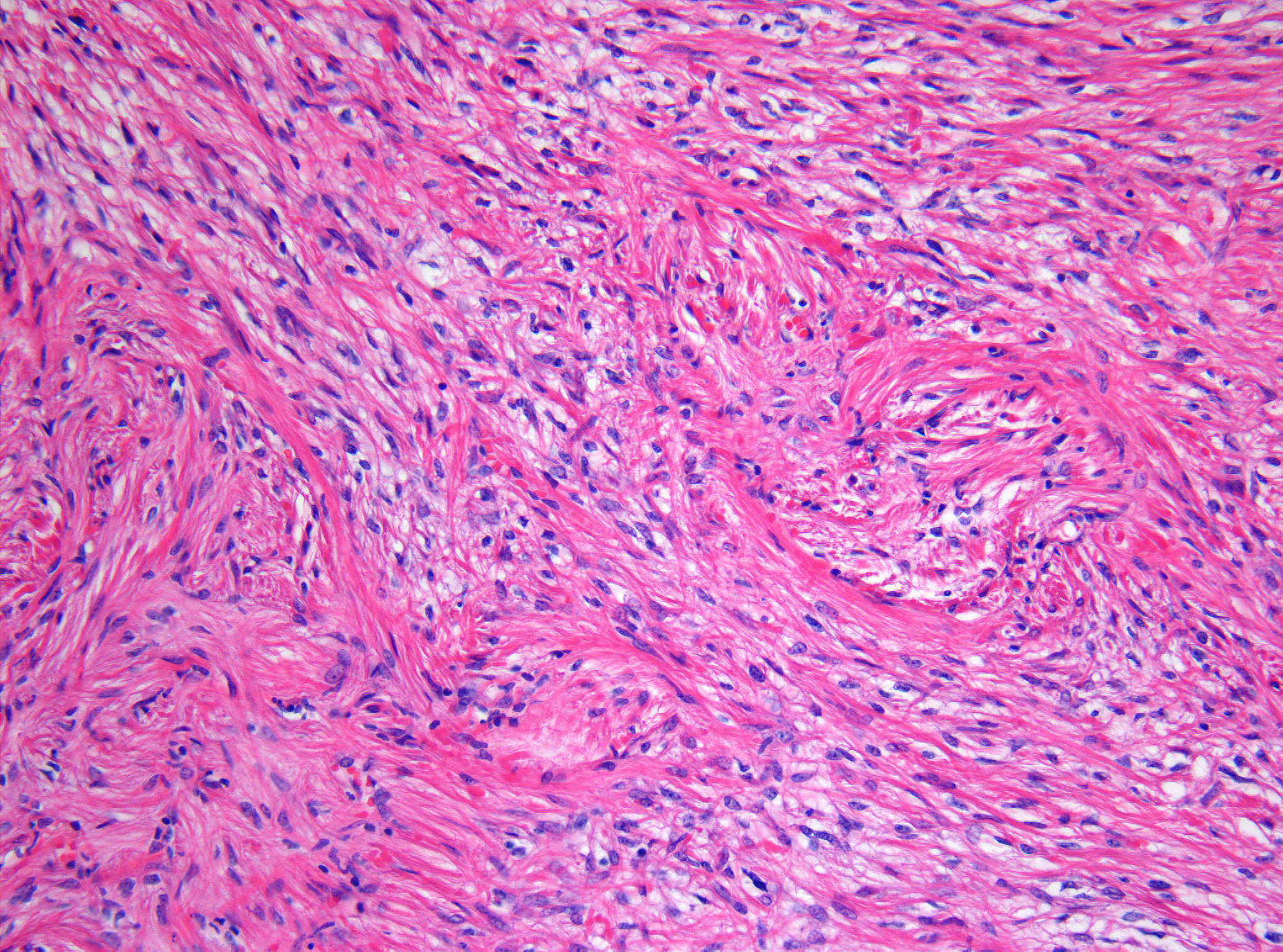
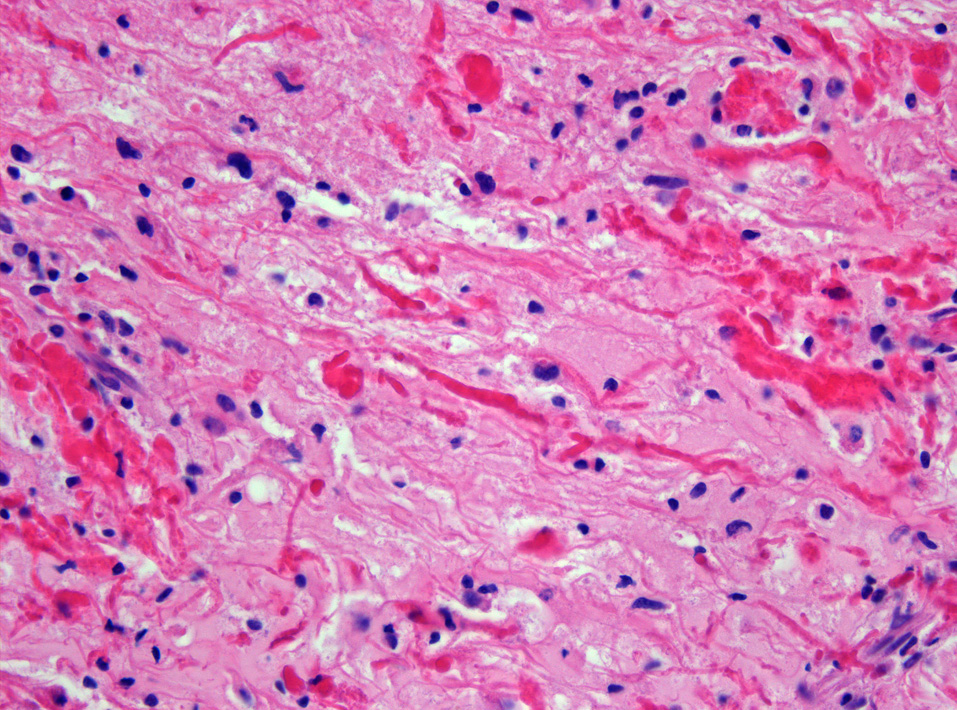
CNS tumours are the most common solid malignancy of childhood and the leading cause of pediatric cancer death. Compared with adult neuro-oncology, the pediatric tumour spectrum is dominated by embryonal tumours, low-grade gliomas (especially pilocytic astrocytoma), ependymomas, and a distinct group of midline and brainstem high-grade gliomas. The 2021 WHO Classification, 5th Edition, formally separates many of these into a 'pediatric-type' category with their own molecular drivers, and integrates molecular information into the diagnostic algorithm of every tumour type.
In children, roughly half of CNS tumours arise in the posterior fossa, with the remainder split between supratentorial and, far less commonly, spinal locations. Tumour type and biology vary strikingly by age, with embryonal tumours (medulloblastoma, ATRT, ETMR) clustering in early childhood and germ cell tumours and craniopharyngiomas more common in older children and adolescents.
References used here
-
WHO Classification of Tumours Editorial Board. WHO Classification of Tumours: Central Nervous System Tumours. 5th Edition (Vol 6 of WHO Classification of Tumours series). IARC / WHO, 2021. ISBN: 978-92-832-4508-7.
-
Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, Hawkins C, Ng HK, Pfister SM, Reifenberger G, Soffietti R, von Deimling A, Ellison DW. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23(8):1231-1251.
-
Albright AL, Pollack IF, Adelson PD. Principles and Practice of Pediatric Neurosurgery. 3rd Edition. Thieme, 2015. ISBN: 978-1-60406-799-6.
-
Winn HR (Editor). Youmans and Winn Neurological Surgery. 8th Edition (4-volume set). Elsevier, 2022. ISBN: 978-0-323-66192-8.